专注全球两三轮车换电一站式解决方案
02083859919 18038691298

当我们都知道哪种电池的电压最高时,现在我们也发现锂硫电池由于其理论能量密度高和硫资源丰富而引起广泛关注。硫化物热解(丙烯腈)(S@pPA)正极可以消除由于固-固转化机制而产生的穿梭效应,但在碳酸盐电解质中存在电解质/电极界面差和动力学迟缓的问题。
通过原位电化学聚合形成的锂硫电池用不燃性聚醚电解质@pPAN,实现了锂硫电池10C超快速充电的超稳定性能,锂硫电池4C400次循环平均库仑效率达到99.9995%。
锂硫电池的介绍
锂硫电池由于其较高的理论能量密度(2600 Whkg-1)和丰富的硫资源,被认为是一种有前途的下一代储能系统。在过去的几十年里,许多涉及锂硫电池所有组成部分的研究已经被报道。
电解液作为阳极和阴极之间的桥梁起着重要作用,对锂硫电池的性能有着至关重要的影响。由于多硫化锂(LiPS)倾向于攻击和破坏碳酸盐电解质,所以醚基电解质经常被用于锂硫电池的电池中,以配合常见的S@C复合阴极的固液溶解沉积机制。
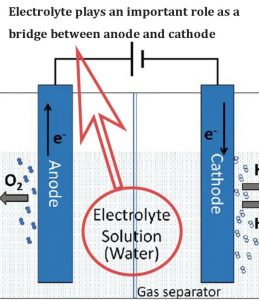
该机制涉及到LiPS不可避免的溶解以实现硫的减少。然而,醚类电解质中的溶解行为表现出典型的穿梭现象,限制了电池的性能,如硫的利用率、循环稳定性和CE。
此外,固液溶解沉积机制严重限制了未来高容量能量密度电池的低孔率和高密度阴极的制造。
在2002年初,硫化物热解聚(丙烯腈)(S@pPAN)被报道在碳酸盐电解质中具有独特的固-固转化机制。与S@C阴极不同,通常采用碳酸盐电解质在阴极表面形成保护性阴极电解质界面(CEI),从而消除了LiPS的溶解和穿梭。
CEI层的质量,包括成分、厚度和机械强度,对电池性能至关重要。然而,在传统的碳酸盐电解质中,由聚碳酸酯组成的CEI显示出厚度增加和大的界面阻抗。同时,不稳定的阴极界面不断消耗锂,导致S@pPAN阴极的CE不足,而这是在实际条件下实现长寿命的一个关键因素。
此外,使用传统碳酸盐电解质的锂金属阳极也存在类似的困境。众所周知,与碳酸盐电解质相比,醚基电解质往往形成较薄的弹性界面层,极化程度较低。
然而,由于高溶解度,LiPS会在普通的醚基电解质中发生溶解。因此,为长寿命的锂硫电池@pPAN电池设计电解质仍然具有挑战性,如图所示。
理想的电解质需要具备以下特性:(1)在锂阳极和硫基阴极上有一个薄而稳定的界面,具有较高的Li+电导率;(2)对LiPS的溶解度低或无溶解度,特别是在筛选醚类溶剂时,(3)不易燃且本质安全的高沸点。
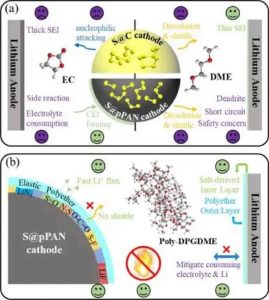
(a)硫基阴极电解质的设计困境的示意图。(b)提议的电解质对两个电极的优越性。
与制约S@pPAN动力学的大阻抗聚碳酸酯CEI相比,聚醚电解质是锂硫电池@pPAN电池的有前途的替代品、
其中,富含聚醚的界面可以明显促进Li+的扩散,这得益于醚类用于锂阳极的优势。此外,聚合物的低LiPS溶解度可能仍然保持S@pPAN独特的固-固转化机制。
此外,原位聚合有利于有效地改善电极/电解质界面,初始的液态单体可以适应体积变化并确保紧密的界面接触。
考虑到醚类溶剂中丰富的ɑ-C-H,ɑ-C-H活化是形成聚醚电解质的可行策略,在电化学环境中重复的电子得失可以作为一种催化剂使用。
快速充电的锂硫电池
二丙二醇二甲醚(DPGDME)是一种无毒、不易燃的醚类溶剂,被用于锂硫电池@pPAN电池。它在循环过程中进行原位电化学聚合,可以有效缓解界面副反应,调控低极化界面相,特别是在阴极一侧。
通过各种表征和分子计算,充分证明了其独特的ɑ-C-H活化机制。本质安全的电解质对锂阳极表现出3000小时的优异稳定性,以及在高温(50℃)和大容量(10 mAh cm-2)下的优异耐受性。
由于所设计的富含聚醚的CEI的快速Li+扩散,锂硫电池@pPAN电池可以实现98.4%的高硫利用率(1645.3 mAh gS-1)和快速充电性能(锂硫电池10C)。此外,它提供了极其稳定的循环,在400次循环中容量保持率为99.5%,平均CE超过99.9995%,表明可忽略不计的不可逆转的锂消耗。
此外,即使在有限的锂和稀释的电解液量的情况下,也能实现优异的性能,这在袋装电池中得到了进一步的证明。

锂硫电池的研究重点
1. 二丙二醇二甲醚(DPGDME)在循环过程中发生原位电化学聚合,可以有效缓解界面副反应,调控低极化界面相,特别是在阴极侧。
2. 该电解液对Li阳极表现出3000小时的优异稳定性,在高温(50℃)和大容量(10 mAh cm-2)下具有良好的耐受性。
3.在400次循环中,容量保持率为99.5%,平均CE超过99.9995%,表明可忽略不计的不可逆锂消耗。
基于DPGDME的电解质的电化学聚合
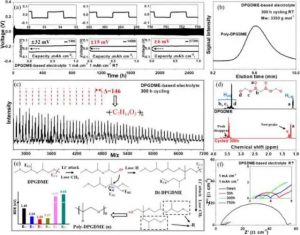
(a)在1 mA cm-2/1 mAh cm-2的条件下,使用基于DPGDME的电解质的Li|Li对称电池的电压变化。(b-d)聚DPGDME的表征:GPC光谱(b),MALDI质谱(c)和1HNMR(d)。(e)聚二甲基硅氧烷的聚集演变。(f) 使用基于DPGDME的电解质的Li|Li对称电池的奈奎斯特图。
二丙二醇二甲醚(DPGDME)是空气污染(NAP)清单上的一种无害溶剂,与锂硫电池中常用的溶剂相比,它保持了优越的特性,包括高沸点、中等介电常数、低密度和粘度。即使在100℃以下,它也能对锂保持出色的化学稳定性。
我们首先制备了基于DPGDME的电解液,并在锂硫电池中进行了测试,以研究其与锂阳极的兼容性,如图所示。 即使在1 mAcm-2的条件下,它也表现出明显的稳定曲线,在2600小时内没有增加过电位(30 mV)。
电压-容量曲线在初始循环时通常是月牙形的,但逐渐演变为先上升后变平的曲线,这与基于聚氧化乙烯(PEO)的电解质相似。这种特殊的分布变化可能导致循环过程中电解质的原地电化学聚合。
此外,上升点逐渐变窄,表明聚合度的增加。为了证明这一点,用凝胶渗透色谱法(GPC)测试了循环后的电解质的分子量。它的重量平均分子量(Mw)很高,接近3350 g mol-1,是DPGDME分子(162 g mol-1)的几十倍,这为电解质的聚合提供了直接证据。

通过基质辅助激光解吸/电离飞行时间质谱(MALDI-TOF)进一步研究了所形成的聚合物结构(聚DPGDME)。如图2c所示,主要分布有一个146 Da的重复单元,对应于(-C7H14O3-)的重复单元。
这表明,在DPGDME(C8H18O3)的聚合过程中,分别去掉了一个-CH3和一个-H。然后跟踪1H核磁共振(NMR)以确认聚DPGDME的具体结构。
纯DPGDME的1H可以分为位于低化学位移(HLS)的非ɑ-H(Ha)和位于高化学位移(HHS)的ɑ-H(Hb、Hc、Hd),摩尔比为1:2。
聚合后,位于HLS的非ɑ-H的比例增加,这意味着-CH3(d)被移除而不是-CH3(a)。何新凤的出现进一步证明了这一点。去掉一边的-CH3(d)后,两个对称的-CH3(a)的周围发生了变化,然后一个-CH3(a)变成了一个新的-CH3(e)的化学位移。
同时,HHS的峰消失了,表明一个ɑ-H被除去,不对称的-CH3(a)/-CH3(e)排除了Ha从静止的-CH3(d)下降的可能性。
与亚甲基上的Hc相比,甲基上的Hb通常显示出更高的活性,并倾向于在攻击下被除去。峰值没有出现在较高的化学位移上,这表明只有Hb而不是Hc下降。峰面积比也证实,在循环过程中,一侧的CH3(d)和一侧的ɑ-Hb分别被除去,形成聚DPGDME。
提出了一种可能的DPGDME聚合机制,如图所示。 在电化学环境中,DPGDME被Li+攻击并产生单O键断裂。产生的DPGDME-自由基将攻击甲基,其中的活性ɑ-H减少,形成二DPGDME。二聚体同时失去-CH3,最终形成多DPGDME。
此外,分子计算提供了进一步的证据,并与核磁共振的结果一致,即具有较低的键解离能(BDEs)的键倾向于断裂。
聚-DPGDME保持着与PEO相似的分子结构,主链和侧链中丰富的单键O-C段可以提供快速的Li+转移,并实现优异的Li+传导性、
这就解释了为什么在不增加过电位的情况下能有稳定的长循环曲线。
此外,如图所示,进一步进行了电化学阻抗光谱(EIS)。即使在750小时的循环后,在聚DPGDME电解液中也观察到稳定的体电阻和极低的界面阻抗,这分别表明电解液的高Li+导电性和薄的有利界面的形成。
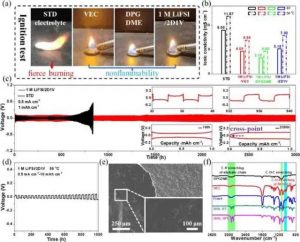
(a) 不同溶剂和电解质的点火试验。(b) 不同电解质的离子传导性。(c,d) 在0.5 mA cm-2/1 mAh cm-2, RT (c), 0.5 mA cm-2/10 mAh cm-2, 50 °C (d)下,锂-锂对称电池的电压演变。(e) 在50℃下循环300小时后的锂沉积的SEM。 (f) 溶剂、初始和循环后电解液的FITR光谱。
此外,选择乙烯-碳酸酯(VEC)作为成膜添加剂,以合理地改变电极界面。

此外,它保持了高沸点和不易燃性,在循环过程中可以发生由金属锂引发的原位聚合。
所有提出的具有不同体积比的DPGDME和VEC(标记为1M LiFSI/nD1V)的电解质都表现出低密度和宽的电化学窗口。
为了验证安全性,进行了直接点火试验,如图所示。所有提议的溶剂和1M LiFSI/2D1V电解液都显示出不可燃性,而在含有10重量%碳酸氟乙烯(STD)的标准碳酸盐电解液中观察到剧烈的燃烧。
即使在高温下浸泡测试1周后,它对锂显示出有竞争力的离子传导性和出色的化学稳定性。
对锂阳极的界面稳定性通过Li-Li电池进一步证实。与STD电解质的持续恶化(<900小时)相比,所有提出的电解质在1600小时内都表现出优异的循环稳定性,而没有明显的过电位增加。值得注意的是,1M LiFSI/1D1V表现出较大的过电位,因为较高的VEC含量在SEI中产生了高电阻的聚碳酸酯。

然而,含有10体积%VEC的电解液表现出较差的循环稳定性,1600小时循环后的电压曲线波动是由于不均匀的锂沉积和树枝状晶体生长造成的,这也是普通乙醚电解液的情况。相比之下,1M LiFSI/2D1V提供了极其稳定的3000小时(125天)的循环,这表明VEC的优化比例在稳健和低阻抗的电极/电解质界面形成中起着重要作用。
此外,在循环过程中观察到了来自交点的特殊轮廓变化,表明两种溶剂的聚合作用增强。在较高的电流密度下,1M LiFSI/2D1V与STD电解质进一步比较,显示出明显的优势。温度升高可以有效加速离子扩散,从而降低过电位,而有害的副反应可能会加剧,特别是在锂阳极和电解质之间。
由于对锂有很好的亲和力,高含量的DDME(2D1V和9D1V)的电解质在50℃下被进一步测试。
值得注意的是,这两种溶剂的不可燃性和高沸点确保了即使在高温下的安全性。拟议的电解质显示出明显减少的10 mV以内的滞后。
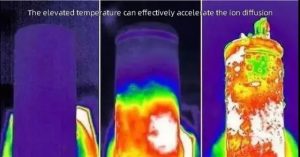
观察到超过800小时的稳定循环,表明即使在高温下也有较少的副反应。此外,大面积容量(>5 mAh cm-2)被认为是该电池走向实际应用的必要因素之一。即使在10 mAh cm-2的严酷面积容量下,所提出的电解质也表现出超过1000小时的长期稳定循环。
在更高的电流密度下,也能实现出色的寿命(>400小时)。对锂阳极界面的扫描电子显微镜(SEM)观察发现,覆盖在锂上的聚合物薄膜更薄,引人注目地显示出致密化和分离。
此外,在剥去一半的薄膜后,内部的沉积层被暴露出来,如图所示是一个光滑的沉积层。
为了证明源于这两种溶剂的聚合过程,在室温和50℃下循环300小时之前和之后,对纯DPGDME、VEC、电解质进一步进行了傅里叶变换红外光谱(FTR)分析。循环后的电池显示没有液体存在,分离器保持透明,表明电解质已经演变成聚合物状态。
如图所示,最初的电解质表现出丰富的脂肪族链单键的H-拉伸峰,这在纯DPGDME中也能观察到。循环后,剩下的峰较少,但变得尖锐,1100eV处对应于单键O-C拉伸的峰显示出强度增加,表明DPGDME在激活和去除ɑ-H后发生了电化学聚合。
此外,RCH=CH双键的C-H单键拉伸峰的消失表明VEC的聚合。源自这两种溶剂的原位聚合可以协同减轻界面副反应和锂的不可逆消耗。
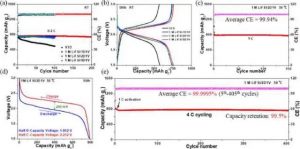
(a)室温下不同电解质的循环稳定性。(b)室温下第50个循环的充/放电曲线。(c)使用1M LiFSI/2D1V在50℃和1℃的循环稳定性。(d) 50℃下第50个循环的充/放电曲线。(e) 在50 °C和4 °C的长周期性能。
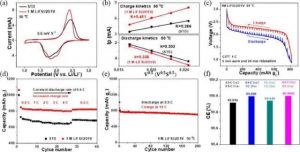
(a) 扫描速率为0.6 mV s-1时的CV曲线。(b) 峰值电流与扫描速率的平方根的线性拟合。(c) GITT曲线。(d) 速率能力。(e) 在10摄氏度下的快速充电性能。 (f) 不同步骤条件下的CE。
进一步评估了提议的电解质对S@pPAN阴极的兼容性。制备了含硫量较高的49.2wt.%的S@pPAN,而不是通常报道的S@pPAN(<45wt.%),目的是提高基于S@pPAN复合材料的比容量(822.6 mAh gC-1)。如图所示,使用不同的电解质对室温下的性能进行了比较。
STD电解质显示出容量的快速下降和过电位的增加。超过45wt.%的过量硫分子参与了与碳酸盐电解质的有害副反应,导致厚的CEI与缓慢的Li+扩散和不可逆转的电解质消耗。
相比之下,所有建议的电解质都表现出更好的循环稳定性和明显降低的过电位,表明在S@pPAN阴极表面形成有利的CEI。
在没有LiPS穿梭的情况下,S@pPAN的固-固转换机制仍然良好。更引人注目的是,与其他电解质相比,1M LiFSI/2D1V的过电位更小,只有315.9mV,半放电容量电压为1.912V。
此外,它在100次循环后实现了1539.5 mAh gS-1或756.8 mAh gC-1(在S@pPAN复合材料中归一化)的显著可逆容量,对应于92.1%的高硫利用率。适度的VEC有利于形成保护性的CEI,将活性材料密封在导电基质中以实现稳定的循环。
同时,主要溶剂DPGDME产生了丰富的聚醚,以构建低阻抗的CEI并促进Li+扩散。它还表现出优异的速率性能,在苛刻的4摄氏度条件下循环300次后,容量保持率达到90.7%。

此外,高温被用来评估1M LiFSI/2D1V的性能。放电/充电曲线显示,由于高温下Li+扩散加速和动力学促进,过电位明显降低。
特别是,第一次放电高原从1.92V上升到2.05V,并在第50个循环时提供了一个较高的半放电容量电压1.951V,电压滞后较低(280mV)。它的比容量明显增加,达到809.5 mAhgC-1(1645.3 mAh gS-1),相当于在50℃时硫的利用率为98.4%。
在1℃下进行100次循环后,可逆容量为791.4 mAhgC-1,每循环的损失率为0.022%,可以忽略不计。此外,使用1M LiFSI/2D1V的电池显示出99.940%的优秀平均CE,表明在每个循环中几乎没有不可逆的锂的消耗。
与室温相比,在高温下实现了高达86.42%的较高能量效率,相应地减少了能量损失。
更重要的是,在初始激活后实现了稳定的长时间循环,在4摄氏度时平均CE为99.9995%,在400次循环中容量保持率为99.5%,效果显著。9D1V和1D1V的电解质也表现出超过99.9%的高CE,而容量相对较低。
高负载的阴极在拟议的电解质中被进一步匹配和测试。它能够实现稳定的循环,在50次循环后具有6.61 mAhcm-2的高平均容量,这优于以前在STD电解质中使用相同粘合剂的报告。
在苛刻的速率下的优异表现表明了快速的Li+传输动力学。循环伏安法(CV)测试表明,1M LiFSI/2D1V在氧化还原峰之间的电位间隔较小,正峰值电流明显较高,几乎是STD电解质的1.5倍,显示出明显增强的快速充电动力学。
在不同的扫描速率下,Li+扩散系数被进一步确定。根据以前的报告,与放电过程相比,STD电解液中的S@pPAN阴极表现出缓慢的充电动力学。
然而,拟议的电解质能够实现惊人的加速充电动力学,甚至优于放电动力学,这意味着出色的快速充电能力。在使用拟议的电解质的锂硫电池的半容量点,也正在进行EIS测试。与放电过程相比,它在充电过程中表现出较低的阻抗。
进一步进行了静电间歇滴定技术(GITT)来研究整个循环过程中的离子传输行为。在充电时的松弛时间内观察到的电压变化比放电时小,表明了超快的充电动力学。
S@pPAN在拟议的电解质中具有诱人的快速充电能力,这一点通过速率容量测试得到进一步证明。在以前的报告中,考虑到缓慢的充电过程是决定性的步骤,采用了增加放电率而保持恒定充电率的方案来评估锂硫电池@pPAN。
在增加充电率的同时增加放电率的相反方案被用来评估快速充电能力。如图所示,与STD电解质较低的容量和增加的极化相比,当充电率从0.5C增加到苛刻的4C时,电解质损失的容量可以忽略不计,并且在返回的速率下容量完全恢复。
图中应用了10C的较高充电率,在200次循环后性能稳定,容量保持率为91.4%。
值得注意的是,CE在锂含量有限的实用锂硫电池中起着关键作用。它是由负极和正极上的界面和副反应的形成引起的不可逆的锂消耗决定的。锂金属阳极上的锂沉积/剥离的可逆性已在许多报道的工作中得到强调。
然而,在评估纽扣电池时,硫基阴极的CE往往被忽视,因为丰富的锂过剩可以补充不可逆的锂耗损,从而消除低CE的影响。作者专注于阴极的锂可逆性,并在不同的步骤中测试CE,包括在0.5C下放电,同时分别在0.5C、1C和2C下充电。
图5将CE在1C下的放电和充电一起进行了比较。研究表明,慢放/快充过程可以达到99.99%以上的CE,这促进了其在锂含量有限的长寿命锂硫电池中的应用。
超高的CE和稳定的循环表明,正极上几乎没有不可逆反应,这归功于CEI的提高和动力学的促进。EIS测试进一步证明了一个稳定的中间阶段,在循环过程中阻抗没有增加。
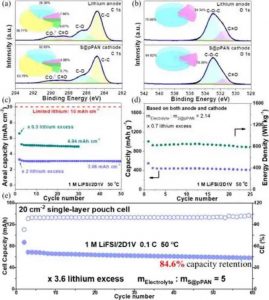
实际条件下锂硫电池@pPAN的电极界面和性能。
(a,b) 锂和S@pPAN的XPS光谱:C1 s(a)和O1 s(b)。(c,d) 有限锂(c)和贫电解质(d)下的性能。(e) 小袋电池的性能。
所设计的电解液尤其与S@pPAN阴极保持了良好的亲和力。循环的锂硫电池@pPAN电池被拆开,用X射线光电子能谱(XPS)分析两个电极,以确认相间的组成。
S、N和F元素来自于LiFSI的分解,它可以构建一个具有良好机械强度和高Li+导电性的改性内层。CEI含量的C 1 s谱在290.1、288.6、286.2和284.8 eV处有四个峰,分别对应于-CO32-、OC=O、CO和单键C。
值得注意的是,主要来自DDME的CEI中含有的C-O单键含量为32.65%,高于来自VEC的-CO32-(4.86%)和OC=O(8.70%)(图6a)。一致的结果显示在O1 s光谱中,单键O-C(533 eV)的强度比C=O(531 eV)高84.62%(图6b)。
大的单键O-C丰度进一步表明,聚醚是在循环过程中从DDME中原地形成的,并呈现出富含聚醚的CEI。锂硫电池阳极上的SEI由类似的成分组成。XPS结果表明,在两个电极的界面上形成了丰富的聚醚。
优化后的CEI超过99.99%的超高CE表明在阴极界面上的不可逆锂消耗可以忽略不计。如图所示,锂硫电池的性能在有限的锂的条件下进行了探索。它显示了在锂过量2倍的情况下超过50次的稳定循环,即使在苛刻的有限锂使用条件下(过量0.5倍)也能提供出色的性能。
此外,电解质的数量被控制以模拟实际锂硫电池的工作条件(2.14克电解质/gS@pPAN,0.7倍的过量锂)。如图所示,它表现出448.4 mAhg-1的初始可逆容量,基于阳极和阴极对应822.5 Whkg-1,超过了STD电解质的性能。
恶劣条件下性能的明显改善以及具有快速充电能力和高CE的动力学,为推动S@pPAN走向实际应用开辟了道路。因此,在有限的锂和可控的电解质下,进一步探索了袋式电池的性能。
提供了746.8 mAhgC-1的高可逆容量,相当于91.2%的硫磺利用率和稳定的循环,60次循环后容量保持率为84.6%。
总结和展望
总之,所提出的电解质在不易燃性、阴极兼容性以及在电化学环境下通过ɑ-C-H活化进行独特的原位电化学聚合方面保持了明显的优势。
与厚碳酸盐衍生的CEI相比,阴极上稳定的富聚醚层不仅可以实现快速的锂扩散和较低的界面阻抗,而且可以缓解锂的不可逆假说。

它以超高的CE(99.9995%)实现了显著的循环稳定性,并表现出显著的快速充电能力(10 C)和增强的充电动力学。
在恶劣的条件下,该软包电池进一步提供了卓越的性能。源自电解质聚合的改性CEI为本质安全和实用的长寿命锂硫电池提供了灵感。如果您想了解软包电池的行业信息,可以参考世界十大软包电池制造商。

电话:02083859919 手机:18038691298
公司地址:广州市黄埔区光谱西路TCL创意产业园530室 邮箱542298629@qq.com
Copyright @ 2026 广州太空人新能源科技有限公司 版权所有 ICP备案:粤ICP备2023005659号-1No:76990
